
FDA-recommended conduct of clinical trials during the COVID-19 pandemic

The Food and Drug Administration (FDA or Agency) has recently acknowledged that the COVID-19 pandemic may have a negative impact on the conduct of clinical trials of medical products. Challenges may arise, for example, from quarantines, site closures, travel limitations, interruptions to the supply chain for the investigational product, or other considerations if site personnel or trial subjects become infected with SARS-CoV-2.
The agency has therefore issued a guidance to assist sponsors in assuring the safety of trial participants, maintaining compliance with good clinical practice and minimizing risks to trial integrity during the coronavirus (COVID-19) pandemic.
The following key points summarize the Agency’s recommendations:
For ongoing trials:
- Ensuring subject safety. Sponsors should consider each circumstance, focusing on the potential impact on the safety of trial participants, and modify study conduct accordingly. Study decisions may include those regarding continuing trial recruitment, continuing use of the investigational product for patients already participating in the trial, and the need to change patient monitoring during the trial. Participants must be kept informed of changes to the study and monitoring plans that could impact them.
Since trial participants may not be able to come to the investigational site for protocol-specified visits, sponsors should evaluate whether alternative methods for safety assessments (e.g., phone contact, virtual visit, continuous remote monitoring, alternative location for assessment, including local labs or imaging centers) could be implemented.
In some cases, trial participants who no longer have access to investigational products or the investigational site may need additional safety monitoring. - Implementing protocol amendments. The need to put new processes in place or to modify existing processes will vary by the protocol and local situation.
Changes in a protocol are typically not implemented before review and approval by the Institutional Review Board (IRB)/Independent Ethics Committee (IEC), and in some cases, by FDA, unless such changes to the protocol or investigational plan aim to minimize or eliminate immediate hazards or to protect the life and well-being of research participants.
The implementation of alternative processes should be consistent with the protocol to the extent possible, and sponsors and clinical investigators should document the reason for any contingency measures implemented. - Documenting missing information. Changes in study visit schedules, missed visits, or patient discontinuations may lead to missing information (e.g., for protocol-specified procedures). It will be important to capture specific information in the case report form that explains the basis of the missing data, including the relationship to COVID-19 for missing protocol-specified information.
- Remote monitoring. If planned on-site monitoring visits are no longer possible, sponsors should consider optimizing use of central and remote monitoring programs to maintain oversight of clinical sites.
- Administration, delivery, and accountability of investigational products. If scheduled visits at clinical sites will be significantly impacted, certain investigational products, such as those that are typically distributed for self-administration, may be amenable to alternative secure delivery methods. For other investigational products that are normally administered in a health care setting, consulting FDA review divisions on plans for alternative administration is recommended.
In all cases, existing regulatory requirements for maintaining investigational product accountability remain and should be addressed and documented. - Efficacy assessments. FDA recommends consultation with the appropriate review division regarding protocol modifications for the collection of efficacy endpoints, such as the use of virtual assessments, delays in assessments, and alternative collection of research-specific specimens, if feasible.
- Data management and statistical analysis plans. If changes in the protocol will lead to amending data management and/or statistical analysis plans, the sponsor should consider doing so in consultation with the applicable FDA review division. Prior to locking the database, sponsors should address in the statistical analysis plan how protocol deviations related to COVID-19 will be handled for the prespecified analyses.
If policies and procedures are not already in place for applicable trials:
- Sponsors, clinical investigators, and IRBs should consider establishing and implementing policy and procedures, or revise existing policy and procedures, to describe approaches to be used to protect trial participants and manage study conduct during a possible disruption of the study as a result of COVID-19 control measures at study sites. Changes to policy and procedures could address, but not be limited to, impact on the informed consent process, study visits and procedures, data collection, study monitoring, adverse event reporting, and changes in investigator(s), site staff, and/or monitor(s) secondary to travel restrictions, quarantine measures, or COVID-19 illness itself. Policy and procedures should be compliant with applicable (regional or national) policy for the management and control of COVID-19.
For all trials that are impacted by the COVID-19 pandemic:
Sponsors should describe in appropriate sections of the clinical study report (or in a separate study-specific document):
- Contingency measures implemented to manage study conduct during the disruption of the study as a result of COVID-19 control measures.
- A listing of all participants affected by the COVID-19 related study disruption by unique subject number identifier and by investigational site, and a description of how the individual’s participation was altered.
- Analyses and corresponding discussions that address the impact of implemented contingency measures (e.g., trial participant discontinuation from investigational product and/or study, alternative procedures used to collect critical safety and/or efficacy data) on the safety and efficacy results reported for the study.

Here, at Empatica, we’re committed to supporting clinical investigators with our AI technology, as/if they need to amend their data management plans and/or monitoring programs during the COVID-19 pandemic.
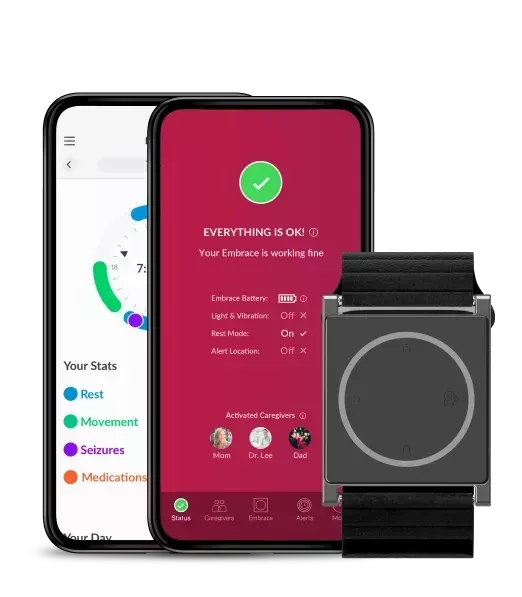
Our Embrace for Research ecosystem is optimized for continuous, clinical-quality observations in an almost real-time and readily visualized format.
The Embrace2 smart watch, designed to be worn 24/7 by the study patients, continuously collects and sends raw physiological and computed data through Bluetooth to the paired Mate app.
Site coordinators, CROs, or researchers are able to access the continuously uploaded and protected sensor data through the Research Portal.
This ecosystem guarantees objective, secure, and remote, real-world data gathering, without the need for physical visits at hospitals/clinics. And it eases the management, scalability, and complexity of virtual trials.
Ultimately, and in line with the FDA’s recommendations, Embrace for Research can contribute to maintaining oversight of clinical sites, minimize risks to trial integrity and offer an alternative method for participants’ safety assessments in trials affected by the COVID-19 pandemic.
If you are interested in using Embrace2 and the Research Portal for your research study/clinical trial, you can get in touch with our team here!
